Dysbindin and Schizophrenia: Itã¢â‚¬â„¢s Dopamine and Glutamate All Over Again.
![]()
Dysbindin-i Interest in the Etiology of Schizophrenia
one
Department of Neuropharmacology and Drug Discovery, Schoolhouse of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China
2
School of Pharmacy Establish for Drug Enquiry, Kinesthesia of Medicine, The Hebrew University of Jerusalem, Jerusalem 91120, State of israel
3
Faculty of Health Sciences, Academy of Macau, Taipa, Macau 999078, China
4
Zhuhai UM Science & Applied science Research Institute, Zhuhai 519080, Mainland china
*
Author to whom correspondence should exist addressed.
Received: 28 August 2017 / Revised: 16 September 2017 / Accustomed: 19 September 2017 / Published: 22 September 2017
Abstruse
Schizophrenia is a major psychiatric disorder that afflicts about 1% of the world's population, falling into the top x medical disorders causing disability. Existing therapeutic strategies accept had express success on cognitive impairment and long-term disability and are burdened by side effects. Although new antipsychotic medications have been launched in the by decades, at that place has been a full general lack of significant innovation. This lack of significant progress in the pharmacotherapy of schizophrenia is a reflection of the complexity and heterogeneity of the disease. To engagement, many susceptibility genes accept been identified to be associated with schizophrenia. DTNBP1 factor, which encodes dysbindin-i, has been linked to schizophrenia in multiple populations. Studies on genetic variations evidence that DTNBP1 modulate prefrontal encephalon functions and psychiatric phenotypes. Dysbindin-1 is enriched in the dorsolateral prefrontal cortex and hippocampus, while postmortem brain studies of individuals with schizophrenia bear witness decreased levels of dysbindin-i mRNA and protein in these brain regions. These studies proposed a strong connection between dysbindin-i part and the pathogenesis of affliction. Dysbindin-i protein was localized at both pre- and post-synaptic sites, where it regulates neurotransmitter release and receptors signaling. Moreover, dysbindin-1 has also been plant to be involved in neuronal development. Reduced expression levels of dysbindin-1 mRNA and protein appear to be common in dysfunctional brain areas of schizophrenic patients. The nowadays review addresses our electric current knowledge of dysbindin-ane with emphasis on its potential role in the schizophrenia pathology. Nosotros propose that dysbindin-1 and its signaling pathways may institute potential therapeutic targets in the therapy of schizophrenia.
i. Introduction
Schizophrenia is an idiopathic mental illness occurring in 0.five–1% of the general population [1]. The clinical symptoms of this disorder include auditory hallucinations, delusions, disorganized spoken communication, abnormal motor behavior, cognitive deficits and other behavioral symptoms [1,2]. Schizophrenia-like symptoms also include negative symptoms, such as reduced response to daily and social activities (motivation), depressive-like emotion or diminished expression of pleasance [three]. These symptoms impair patients' daily performance, and tin can be disabling [4]. Despite the efforts to develop constructive interventions, drugs and psychosocial therapies, an efficient handling of schizoaffective disorder is non yet available. Most of the antipsychotic drugs control some schizophrenic symptoms by affecting the dopamine and/or serotonin in the brain, even so schizophrenia requires chronic therapy and antipsychotics usually cause intolerable side effects [v]. Failures of investigational new drugs for schizophrenia have left huge unmet medical needs for patients. Given the recent lackluster results, it is imperative that new drug candidates that target pathophysiological alterations that are shared in specific patient populations is becoming increasingly necessary for hereafter investigational new drugs [6].
It is more often than not accepted that genetic and environmental factors can trigger the affliction [7]. In fact, cistron-past-environment interactions and epigenetic alterations modified by environmental and social factors are now considered as possible facets of the schizophrenia etiology [8]. A large number of epidemiological studies carried out in family and twin established that the vulnerability to develop schizophrenia is largely genetic in stratified patient populations [ix,x]. Family studies constitute that the lifetime morbid risks of schizophrenia in relatives of patients is ten times higher than that in relatives of controls [11,12]. Heritability of schizophrenia is around fourscore–85% and twin studies have indicated 50% risk in monozygotic and 17% chance in dizygotic twin obtaining the diagnosis (if the other twin already has it) [12,13]. It is interesting that the rates of developing schizophrenia were quite low and no deviation was observed in the adoptive families of both affected and control groups [12]. These studies clearly indicate the important role of genetic factors in the pathogenesis of schizophrenia. The genetic basis of schizophrenia is complex and genome-wide association studies (GWAS) have identified hundreds of single nucleotide polymorphisms (SNPs) associated with schizophrenia [14].
Many studies take shown that dystrobrevin binding protein-ane (dysbindin-1) is one of the important potential susceptibility genes for schizophrenia [15,16,17,18,19,20,21]. Accumulating evidence shows that the level of dysbindin-1 is reduced in postmortem brains from schizophrenia patients [22,23]. Studies on its neurobiological functions indicate that dysbindin-ane regulates neurotransmitter release, postal service-synaptic receptor expression and encephalon development [24,25,26,27,28,29]. Thus, an enhanced understanding of the biological functions and molecular pathways mediated by dysbindin-i is required to better exploit the therapeutic potential of dysbindin-ane for the treatment of schizophrenia. In this review, we volition examine the electric current understanding and bear witness that are proposing dysbindin-1 involvement in schizophrenia and explore its potential as an intervention target for the treatment of schizophrenia.
2. Expression of Dysbindin-i in the Brain and Its Biological Functions
Dysbindin-1 is a protein encoded past dystrobrevin-binding protein 1 gene (DTNBP1), which is located on the short (p) arm of chromosome 6 at position 22.3 [30]. Initially, dysbindin-one was found to be a component of the dystrophin-associated protein circuitous (DPC) in skeletal muscle cells. It is believed that dystrophin-1, which links the jail cell cytoskeleton to the extracellular matrix, contributes to the stability of muscle fibers and, therefore, the loss of dysbindin-1 is probably involved in the pathology of muscular dystrophy [31,32]. DPC is also highly expressed in the encephalon, in particular the cortex and the hippocampus. As a component of the DPC circuitous, dysbindin-1 in the primal nervous system (CNS) maintains the structure and physical stabilization of neuronal synaptic membrane [33].
Dysbindin-ane is also a role of biogenesis of lysosome-related organelles circuitous one (BLOC-one) [34], which is involved in the biogenesis of specific components, such as melanosomes and platelet-dense granules, of the endosomal-lysosomal arrangement [35]. In the CNS, BLOC-1 subunits co-localized with synaptic vesicles and synaptosomes derived from synaptic endings [36]; therefore, it was proposed that these BLOC-1 subunits command membrane expression and lysosomal commitment of post-synaptic receptors [37]. Every bit a component of BLOC-ane, dysbindin-ane is establish primarily in axon or synaptic terminals in the striatum, neocortex, cerebellum and hippocampus [32], brain areas afflicted in schizophrenic patients. BLOC-1 complex contains many proteins, including pallidin, muted, cappuccino, dysbindin, snapin, blosl, blos2 and blos3 [38]. Mutation or deletion of dysbindin-one subunit is associated with a destabilization of these proteins of the BLOC-1 complex [37], and defects on the dysbindin-1 complex contribute to synaptic and excursion deficits [36]. Moreover, its deficiency affects the expression of post-synaptic neurotransmitter receptors [37,39,40,41], which are involved in schizophrenia pathogenesis.
Dysbindin-ane is an evolutionary conserved protein composed of approximately 350 amino acids and containing two coiled-gyre domains [42]. In that location are three dysbindin-1 isoforms, namely dysbindin-1A, -1B and -1C [42]. All 3 of these isoforms are highly expressed in neuronal cells, while dysbindin-1A is the longest and major isoform expressed in the brain [28]. Dysbindin-1A and -1B accept the aforementioned North-termini, whereas dysbindin-1B lacks exon encoding the PEST ((proline (P), glutamic acid (E), serine (Southward) and threonine (T)) domain in the C-terminal [43]. Dysbindin-1A and -1C have the same C-termini, while dysbindin-1C lacks the N-terminal 81 amino acids [43]. Isoforms 1A and 1B are mainly localized in nucleus, whereas the isoform C is exclusively expressed in the cytosol [42]. Dysbindin-1A is expressed on postsynaptic densities (PSDs), dysbindin-1B is mainly expressed in synaptic vesicles, and dysbindin-1C is expressed both in synaptic vesicles and present in PSDs [28]. Knock down of dysbindin-1 resulted in an imbalance of the dopaminergic system and dysregulation of hippocampal synaptic transmission [26,27,44,45,46], pathological processes of schizophrenia. The levels of both dysbindin-1B and 1C are reduced in the hippocampus of schizophrenic patients, while level of synaptic dysbindin-1A was not afflicted [36], suggesting that different dysbindin-i isoforms have different biological functions in schizophrenia. Among the three isoforms, dysbindin-1C was implicated in neurogenesis and neurodevelopment. Deficiency of dysbindin-1C leads to a reduction in mossy cells and delayed maturation of newborn neurons in the adult hippocampus [29]. These data suggest that singled-out dysbindin-i isoforms regulate neurodevelopment and reduced expression dysbindin-1C is probably associated with impairment of adult hippocampal neurogenesis.
3. Interaction of Dysbindin-1 with Cellular Proteins
Information technology is widely accepted that that dysbindin-1 interacts with multiple proteins and exhibits different biological functions in different tissues [32,47]. In the CNS, dysbindin-1 and DPC were implicated in the formation and stability of neuronal synapses likewise as the regulation of dendritic spine morphogenesis [33]. Schizophrenia is commonly viewed every bit a neurodevelopmental disorder originating from decreased spine density and dumb synaptic connectivity [48]. Therefore, it is hypothesized that deficiency of dysbindin-1 leads to profound dysfunction in synaptic connectivity, and eventually contributes to the schizophrenia-like pathology [33]. Too the originally identified interacting poly peptide dystrobrevin, it has been reported that dysbindin-1 interacts with many proteins, such equally histone deacetylase 3 (HDAC3) [49], DNA-dependent protein kinase (DNA-PK) [42], nuclear factor-kappa B (NF-κB) [50], disrupted in schizophrenia 1 (DISC1) [51] and snapin [52].
Dysbindin-1 formed a poly peptide complex with HDAC3 in human neuroblastoma cells and in mouse encephalon. The interaction between dysbindin-ane and HDAC3 occurred in an isoform-specific manner: HDAC3 coupled with dysbindin-1A and -1B, just not -1C. It was besides found that dysbindin-1B expression was increased in the nucleus in the presence of HDAC3, and conversely, that the phosphorylation level of HDAC3 increased in the presence of dysbindin-1B [49]. Therefore, it is tempting to propose that dysbindin-ane may regulate gene transcription through an interaction with HDAC3. In patients with psychiatric disorders, histone acetylation is significantly reduced and inhibition of HDAC might exist a promising strategy for noesis improvement in schizophrenic patients [53]. In the nucleus, dysbindin-1 forms a protein complex with HDAC3 and Deoxyribonucleic acid-dependent protein kinase (DNA-PK). DNA-PK circuitous promotes the phosphorylation of both dysbindin-1 and HDAC3 [42]. Among the three isoforms, dysbindin-1A, and -1B localize in the nucleus and collaborate with HDAC3, while but dysbindin-1B facilitates the phosphorylation of HDAC3 by Deoxyribonucleic acid-PK [49]. Since dysbindin-1A and -1B have different C-termini, it is hypothesized that C-terminal plays a key role in mediating the isoform-specific interaction with HDAC3 [49]. NF-κB is a transcription factor involved in neuronal outgrowth and synaptic plasticity [54]. In schizophrenic patients, the activity of NF-κB is significantly decreased [55], implicating NF-κB in the etiology of schizophrenia. Furthermore, dysbindin-1A is degraded in the nucleus via the ubiquitin-proteasome system and dysbindin-1A amino acids 2-41 at the North-terminus are required for this process [50]. Dysbindin-1A has been proved to interact with p65, a subunit of NF-κB, in the nucleus and enhance the transcriptional activity of NF-κB [50]. Considering nuclear-cytoplasmic shuttling property in combination with its nuclear degradation and possible regulation of NF-kappa B activities, it is reasonable to propose an important function for dysbindin-1A during schizophrenia pathogenesis. Moreover, as NF-κB has been linked to neuroinflammatory responses in relation to neurodegeneration [56] and schizophrenia [57], we suggest that dysbindin-1A interaction with the NF-κB signaling cascade is responsible for localized neuroinflammation in college areas of the brain involved in the behavioral and clinical symptoms of schizophrenia.
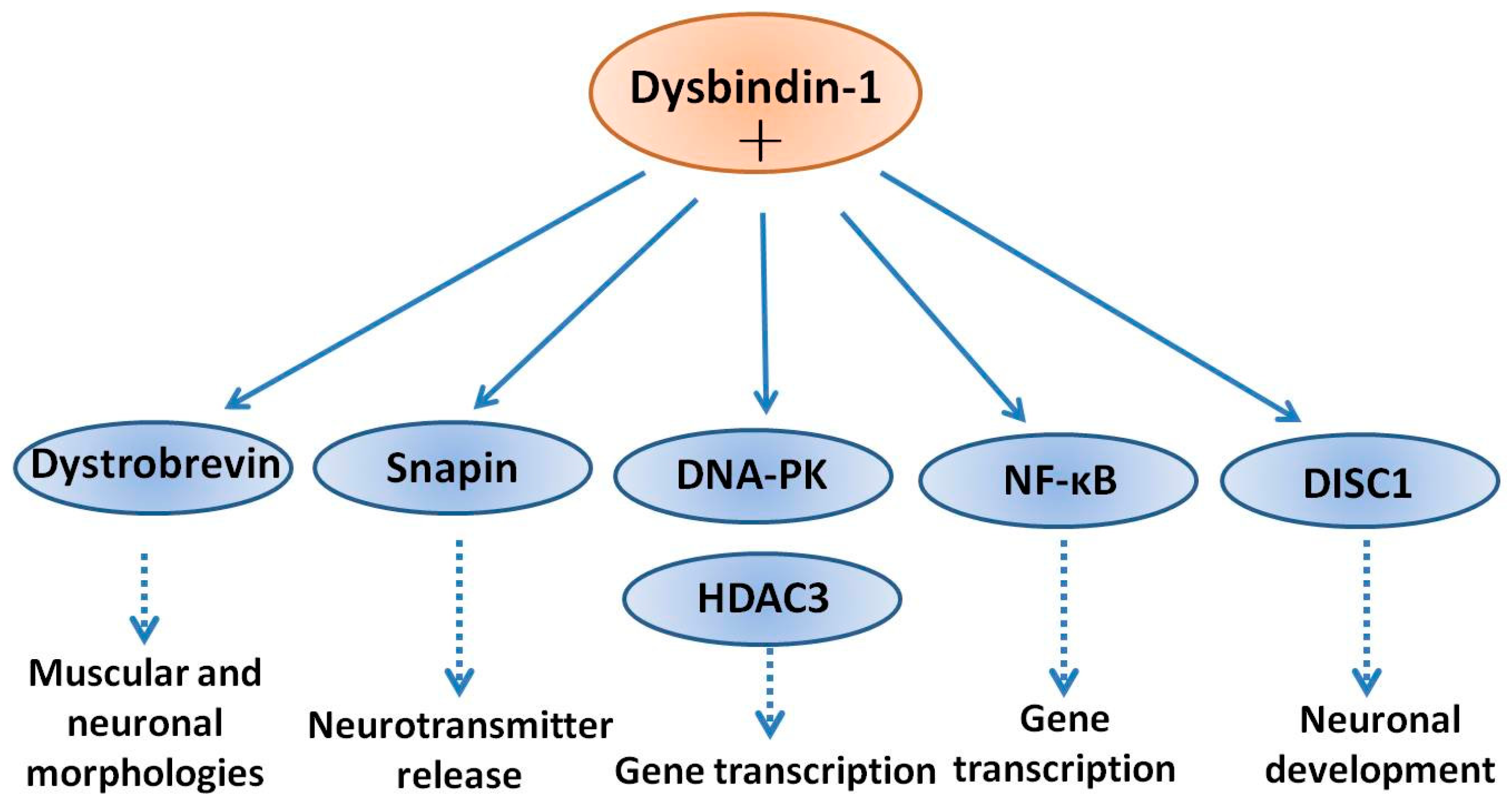
DISC1 is another schizophrenia susceptibility factor playing roles in neuronal evolution [58]. In neuronal cells, DISC1 forms a complex with dysbindin-1, increases its stability in association with a reduction in ubiquitination and the physical interaction is critical for the process of neurite outgrowth. Furthermore, knockdown of DISC1 or expression of a deletion mutant, effectively decreased the levels of endogenous dysbindin-one. Moreover, the neurite outgrowth defect induced by knockdown of DISC1 was partially reversed by co-expression of dysbindin-1. Taken together, these results bespeak that dysbindin-1 and DISC1 class a physiologically functional circuitous that is essential for neurite outgrowth [59]. Snapin is a binding partner of dysbindin-1 in the brain. Tissue fractionation of whole mouse brains and human hippocampal formations revealed that both dysbindin-1 and snapin are full-bodied in tissue enriched in synaptic vesicle membranes and less commonly in postsynaptic densities. Consistent with that finding, localization studies indicated that dysbindin-i is located in: (i) synaptic vesicles of axonal terminals in the dentate gyrus inner molecular layer and CA1 striatum radiatum; and (two) postsynaptic densities and microtubules of the dentate neurons and CA1 pyramidal cells [60]. The office of dysbindin-one in presynaptic, postsynaptic and microtubule locations may all be related to known functions of snapin in regulation of neurotransmitter release [61]. Dysbindin-1 and snapin are components of BLOC-1 and snapin is some other binding partner of dysbindin-ane in vitro and in the brain [threescore]. Deletion in the DTNBP-1 factor causes reduced level of snapin, which is accompanied by defects of synaptic morphology in hippocampal neurons and schizophrenia-like behaviors in mice [62]. Disruption of dysbindin–snapin complex is supposed to affect microtubule associates, which could contribute to reductions in the neuronal cell size and reduced dendritic density. All of these morphological alterations were oft observed in schizophrenia [sixty]. A scheme of the above interactions of dysbindin-1 with several neuronal proteins is shown in Figure 1.
4. Clan of Dysbindin-one with Schizophrenia
Numerous studies have suggested a genetic predisposition to schizophrenia, and many genes, including DISC1, catechol-O-methyltransferase (COMT), neuregulin 1 (NRG1), and DTNBP1, have been identified equally candidate susceptibility genes [2,63]. DTNBP1 is found at chromosomal locus 6p22.three and mutations on this locus have been linked to schizophrenia [64]. Several single SNPs of DTNBP1 were suggested to influence multiple psychiatric phenotypes in schizophrenic patients. For example: (i) SNP rs1997679 and SNP rs9370822 were proven to be associated with visual hallucination [65]; (2) SNP rs4236167 was associated with auditory hallucination [65]; (three) SNP rs9370822 and SNP rs9370822 were establish associated with olfactory hallucinations [65]; (4) SNP rs909706, rs760761 and rs1018381 were associated with attention [66,67]; (5) SNP rs2619522 was correlated with hippocampal and prefrontal greyness matter volumes in schizophrenic patients [68]; and (six) SNP rs9370822 affected glutamatergic or dopaminergic neurotransmission and has been found to be associated with a number of psychiatric weather including schizophrenia [69]. In 2014, Schizophrenia Working Group of the Psychiatric Genomics Consortium reported a multi-phase schizophrenia genome-broad clan study of more than than 150,000 people and found 108 schizophrenia-associated genetic loci [seventy]. Many of these were involved in dopamine receptor subtype two (D2R) and glutamatergic neurotransmission, findings consistent with pathophysiological hypotheses of schizophrenia [70]. Interestingly, there are several works reporting in fauna studies that genetic disruption of dysbindin-i can alter dopaminergic and glutamatergic neurotransmission [46,71,72,73,74]. Therefore, mutations in Dysbindin gene causing partial or full loss of role, may correspond as a direct genetic bridge between these two neurotransmitter systems [74]. Statistical evidence for genetic association is strongly supported by altered DTNBP1 gene expression in schizophrenic encephalon.
Postmortem studies have indicated that mRNA and poly peptide expression of dysbindin-1 is decreased in the brains of schizophrenic patients [75]. Talbot et al. found that presynaptic dysbindin-1 was significantly reduced in the hippocampal germination in schizophrenic populations [22]. The reduction of dysbindin-1 poly peptide was related to glutamatergic alterations and was proposed to contribute to the cerebral deficits in schizophrenia [22]. Weickert et al. constitute that patients with schizophrenia had a meaning reduction of dysbindin mRNA levels in the dorsolateral prefrontal cortex and midbrain [23]. Therefore, because the reduced dysbindin-1 expression in schizophrenic patients, together with the data from SNPs of DTNBP1 cistron mutations, we propose that dysbindin-1 is an etiologic cistron in schizophrenia.
5. Dysbindin-1 Mutation Links to Schizophrenia-Similar Behaviors
Dysbindin-1 knockout mice offer an ideal tool to written report the biological and pathological roles of dysbindin-1 in the brain and development of schizophrenia. Sandy (sdy) mice are dysbindin-1 knockout mice generated in the Jackson Laboratory. These mice have a deletion mutation occurring spontaneously in the inbred DBA/2J mice on the cistron encoding dysbindin-1 (DTNBP1) [76]. Dysbindin-1 deletion has no outcome on body weight, appearance, sensory-motor reflexes and neuromuscular force, while Sdy mice displayed decreased locomotor activeness and deficits in social interaction [44]. This phenotype is peradventure due to a decreased motivation to explore, which is highly related to the negative symptoms of schizophrenia. In add-on to hypo-locomotor activity, Sdy mice as well showed cognitive losses, including arrears of long-term memory retentivity and dumb working retentivity [77], representative neurobiological traits observed in patients with schizophrenia [78]. The original Sdy mice were based on DBA/2J genetic groundwork. This groundwork itself is characterized past locomotor and memory deficits that may confound the explanation of phenotypes observed in this mouse [79]. Hence, the Sdy mutant mice were thereby produced from the C57BL/6J groundwork (dys−/−). Consistently, dys−/− mice also showed displayed clear deficits in spatial learning and memory using the Morris water maze and T-maze tests [46,71]. Still, dys−/− mice are hyperactive in an open up-field test [71]. In improver, aberrant pre-pulse inhibition (PPI) of an acoustic startle stimulus is usually observed in individuals with schizophrenia [80]. Like to this observation, dys−/− mice showed higher acoustic startle reactivity to the 120-dB stimulus [46], indicating that reduced dysbindin-one levels in mice, mimicked the increased reactivity to stressful events seen in patients. These results support the clan of dysbindin-1 to psychosis in humans. Mechanistic studies showed that dysbindin-1 mutation in mice dysregulated pre- and post-synaptic glutamatergic transmission and the expression of the North-methyl-d-aspartic acid (NMDA) receptors was significantly decreased [81]. Dys−/− mice also revealed deletion of dysbindin-ane, reduced expression of Catwo+/calmodulin-dependent protein kinase II (CaMKII) in medial prefrontal cortex [46] and enhanced cell surface recycling and insertion of D2R into the jail cell membrane [41], processes which may strengthen D2R-mediated signaling. Collectively, the results from dysbindin-ane knockout animals back up the view that dysbindin-1 may increment the run a risk for schizophrenia by disrupting glutamate and dopamine-related mechanisms regulating cortical function and neuronal excitability.
6. Regulation past Dysbindin-1 of Neurotransmitter Receptors
The dysfunction of dopamine is a well-established working hypothesis of schizophrenia [82]. Recently, a "dual topographic dysregulation" of dopamine alteration has been proposed in the reevaluation of the DA hypothesis of schizophrenia [82,83]. While within the striatum, peculiarly in the rostral caudate, the release and synthesis of dopamine is excessive, exterior of the striatum, the release of dopamine is in deficit in nigh brain regions, (cortex, hippocampus, and midbrain) [82,83]. In addition, striatal-cortical connections are significantly disrupted in patients with schizophrenia. In this context is important to stress that abnormal striatal connectivity specifically correlates with severity of positive symptoms and lower density of extra-striatal D2R within the same private [84]. On the other hand, cumulative evidences have shown that the core pathophysiology of schizophrenia might involve dysfunction of glutamate [two]. Summarizing, increasing molecular evidences back up a dual part of dysbindin-i in dopamine and glutamate signaling.
Several studies have examined the regulatory role of dysbindin-1 in dopaminergic manual: (i) dysbindin-1 is a component of BLOC-1 involved in intracellular protein trafficking and synaptic homeostasis [39]; (2) mutation of dysbindin-1 caused impaired trafficking of D2R and increased expression of D2R on cell surface of brain cortical neurons [41]; (iii) decreased expression of dysbindin-ane did not touch on dopamine D1 receptors (D1R) [41]; (4) decreased DTNBP1 mRNA using siRNA transfection increased the expression of D2R in both SH-SY5Y neuroblastoma cells and in chief cultured cortical neurons [41]; and (5) in dysbindin-1-deficient mice, pyramidal neurons in medial prefrontal cortex were more than sensitive to D2R agonist-induced behaviors, while they were less sensitive to D2R antagonist [46].
These findings indicate that dysbindin-1 regulates the expression of D2R on neuronal cell surface too as modulates D2R-related behaviors. Moreover, dysbindin-one likewise affects D2R-mediated signaling. Dysbindin-one deficiency in the brain reduced Ca2+/calmodulin-dependent protein kinase Two (CaMKII) expression and signaling in medial prefrontal cortex, while chronic D2R agonist treatment reversed the changes in signaling [46]. Additionally, Dysbindin-1 reduced dopamine-induced adenylate cylase/cAMP signaling and phosphorylation of poly peptide kinase B/glycogen synthase kinase-3β (Akt/GSK3β) and extracellular signal-regulated kinase1/2 (ERK ane/2) [85]. Hence, reduced expression of dysbindin-1 in the encephalon of schizophrenic patients may subtract dopaminergic signaling, supporting its link to the etiology of schizophrenia.
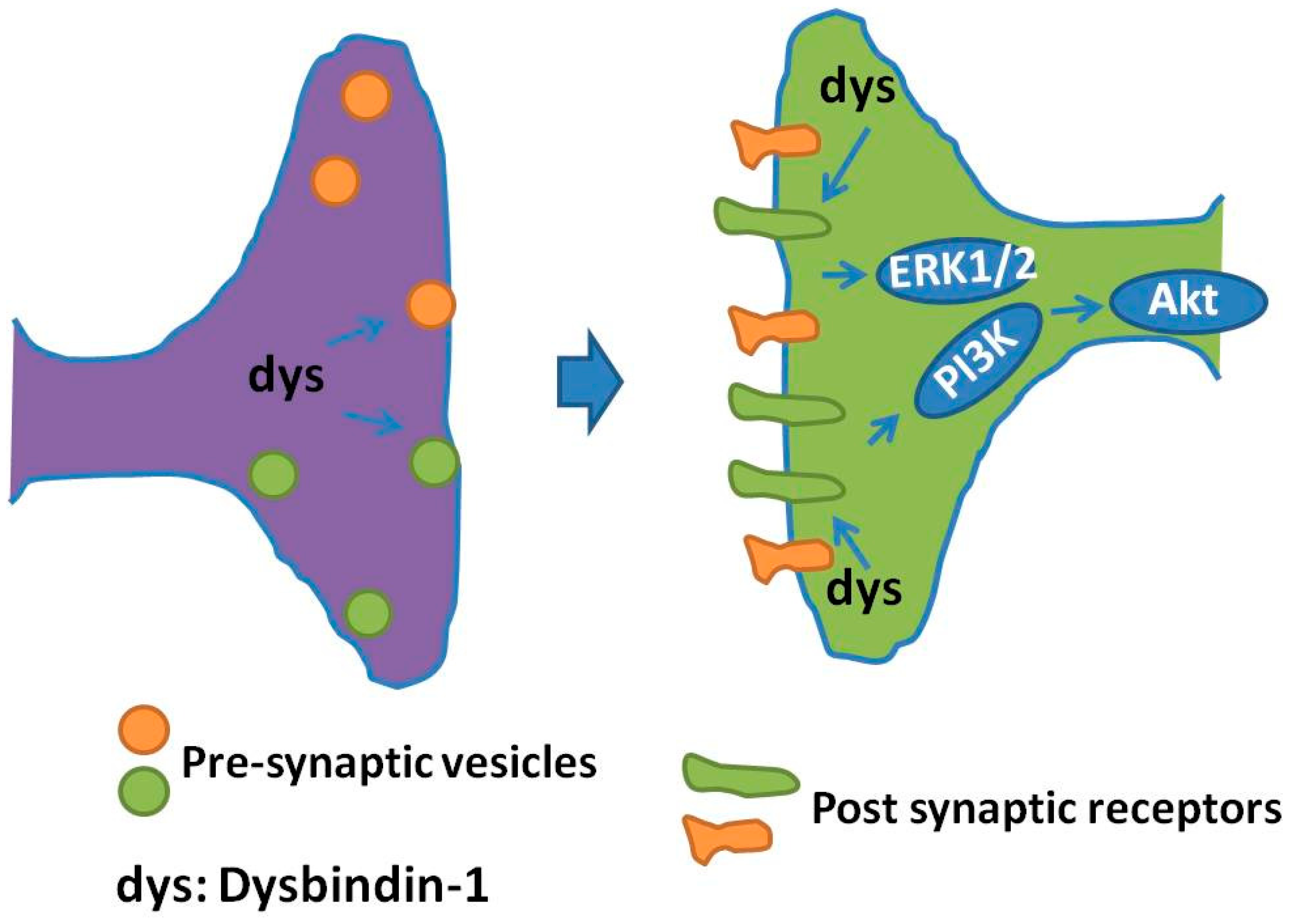
Besides dopamine receptors, dysbindin-1 may touch on glutamate receptor(south) and their signaling: (i) decreased NMDA-evoked currents and NMDA receptor subunit i expression was observed in prefrontal pyramidal neurons in dysbindin-1 mutant mice [81], indicating that dysbindin-one deficiency downward regulates both the expression and part of NMDA receptors; (ii) hippocampal slices from dysbindin mice exhibited an enhanced long term synaptic potentiation (LTP), a process straight correlated with elevated neuronal prison cell surface expression of NMDA type subunit 2A [86]; (iii) dysbindin-1 deficiency acquired impairment in hippocampal synaptic plasticity and hippocampal-dependent memory [87], functions in which both mGluRI and NMDA receptor are involved [73]; and (4) dysbindin-1 is expressed in forebrain glutamatergic neurons and dysbindin mutants showed impairments of prefrontal cortical glutamatergic circuits [88]. Cumulatively, these findings propose a part for dysbindin-ane in NMDA system, which is supposed to affect cerebral impairments of schizophrenic patients [87]. A schematic of the regulation of pre-synaptic vesicles and post-synaptic receptors regulated by dysbindin-1 is presented in Figure 2.
7. Dysbindin-1 Regulation of Neurite Outgrowth
Neurodevelopmental disturbance may contribute to the pathogenesis of schizophrenia and schizophrenia had as well been viewed as a developmental encephalopathy [89]. This hypothesis is farther supported by the fact that many of candidate genes in schizophrenia have also been clearly shown to exist linked to the neurodevelopment procedure [89]. The sprouting and elongation of neurites (neurite outgrowth) is an initial critical process in the early phase of neurodevelopment of the nervous system [ninety]. Dysbindin is expressed embryonically and the level of dysbindin is higher during embryonic and early on postnatal stage than that in adulthood [24]. The average level of dysbindin-one in hippocampus and cerebral cortex from postnatal mean solar day ane mice was three times higher than that from postnatal 45 days aged mice [24]. The role of dysbindin-i in neurite outgrowth has been extensively documented. Main cultures of dysbindin-1-deficient neurons display neurite extension defects: reduced number of neurites and shorter length compared with wild type neurons [24]. In line with these findings, knockdown of dysbindin-one in rat hippocampal neurons using siRNA caused abnormally elongated, immature-dendritic protrusions [33]. Consequent with these findings, siRNA-mediated knockdown of dysbindin-1 in human neuroblastoma SH-SY-5Y cells, caused shorter neurites and abnormal arrangement of the actin cytoskeleton at their growth cone [91]. Furthermore, in vitro cultured hippocampal neurons, derived from dysbind-1 knockout mice, showed morphological abnormalities of the actin cytoskeleton on growth cones [91]. Together, these results point that deficiency of dysbindin-1 might cause subtle defects during neurodevelopment, neural network organization, and activity. Dysbindin-1 may as well touch on pathologically neurite outgrowth through interacting with other proteins. For example, both dysbindin-ane and DISC1 are susceptibility factors for schizophrenia [two,59]. DISC1 has been reported to be an essential component regulating neurite outgrowth during neuronal differentiation [92]. In the CNS, DISC1 forms a complex with dysbindin-1 and their physical interaction is essential for normal neurite outgrowth [59]. Moreover, dysbindin-i may also facilitate neurite outgrowth indirectly through regulating the transcriptional activity of p53 [25], which is a tumor suppressor and involved in neurodevelopment likewise [93]. These studies stress the of import role of dysbindin-one in the procedure of neurite outgrowth. Therefore, further studies are needed to shed light on interactions between signaling pathways regulating neurite outgrowth and dysbindin-1.
8. Dysbindin-1 Is Required for Diverse Presynaptic and Postsynaptic Mechanisms
Since dysregulation of presynaptic and postsynaptic mechanisms of neurotransmitter release could contribute to the etiology of schizophrenia [94], defining the protein interaction networks for dysbindin-1 in the neuronal synapse will help understanding its functions. Dysbindin-i is localized in the synapse and has multiple essential roles in presynaptic and postsynaptic pharmacology [39]. In recent years, using biochemical methods such every bit immunoprecipitation, mass spectrometry and protein expression analysis, a number of interactions betwixt dysbindin-1 and an array of other neuronal proteins were identified. In the presynaptic terminal, dysbindin-1 is involved in unlike aspects of vesicle trafficking processes [95]. For example, dysbindin-ane participates in synaptic vesicle biogenesis and cargo sorting through interacting with BLOC1 and adaptor-related protein circuitous-3 (AP3) [95], dysbindin-1 regulates vesicle trafficking through interaction with actin and tubulin-based cytoskeletons, such as dynactin and tubulin/actin proteins [95]. Moreover, dysbindin-1 is involved in membrane targeting and vesicle tethering by bounden and interacting with exocyst [95]. At pre-synaptic terminals, dysbindin is co-localized with Munc18-ane, a neuron-specific protein essential for the exocytosis of synaptic vesicles [96], modulating synaptic vesicle fusion and neurotransmitter release [97]. These findings suggest that impairment of the presynaptic vesicle functions regulated by dysbindin-1 may be a pathogenic mechanism in schizophrenia.
Recently, Gokhale et al. applied quantitative mass spectrometry to identify the proteomics of neuronal cells with dysbindin-1 deficiency [98]. Both dysbindin-1 and actin-related protein 2/iii (Arp2/three) circuitous subunits localized to presynaptic and postsynaptic terminals in neuronal cells [60,98]. It is known that synaptic terminals' dysbindin and Arp2/three complex are involved in regulation of structural plasticity of dendritic spines, actin protein polymerization, and expression of neurotransmitter receptors [99,100,101]. Since the expression of Arp2/3 complex is reduced in dysbindin-deficient cells, it is assumed that interaction of dysbindin-1 with the Arp2/three complex modulates presynaptic plasticity and adaptive synaptic responses [98]. These findings propose that dysbindin-one is necessary for neuronal synaptic plasticity. A proteome-wide search for expression of proteins which are affected past dysbindin/BLOC-1 deficiency in neuronal cells showed that expression of 224 proteins is altered [102]. Annotation of these proteins to neuronal functions indicates that in a majority they are involved in neurotransmitter vesicle fusion and synaptic plasticity [102]. Additional quantitative proteomic studies have identified changes in expression of proteins and polypeptides sensitive to dysbindin/BLOC-i loss of function, including: (i) the BLOC-1 subunits, such as Bloc1s1-5 and snapin; (two) dynactin circuitous, such as alpha-centractin and dynactin 2; (3) exocyst complex, such as exocyst 3 and exocyst 4; (iv) tubulin/actin associated proteins, such as actin alpha 1 and tubulin blastoff 1b; (five) AP3 complex, such every bit adaptor-related protein complex-3B1 and -ii; (vi) vesicular send/trafficking associated/fusion apparatus, such as adaptor protein 2A1, the vesicle associated membrane protein 7, syntaxin-binding protein one and 5; (vii) proteasome subunits, such equally proteasome modulator 9 and proteasome subunit blastoff type iv; and (viii) other proteins, such every bit the copper-transporting P-type adenosine triphosphatase (ATP7A), the N-ethymaleimide-sensitive gene, annexin A2, syntaxin seven and 17, synaptosomal-associated protein 25 and family unit with sequence similarity 91 member A1 [95,98,102,103,104]. Hereafter investigations on these dysbindin-1-interacting proteins are expected to expand dysbindin-i neuronal functions and provide culling, additional molecular targets for schizophrenia susceptibility.
9. Summary, Conclusions and Perspective
Electric current studies take suggested that genetic factors contribute to the evolution of schizophrenia and dysbindin-1 has been identified as one of the susceptibility genes [49]. The post-obit accumulating evidences suggest the contribution of dysbindin-1 to the pathogenesis of schizophrenia: (i) dysbindin-1 is highly expressed in the dorsolateral prefrontal cortex and hippocampus [28], identified as major regions that may be altered in schizophrenic patients [105]; (ii) multiple SNPs of DTNBP1 are suggested to influence different psychiatric phenotypes [65,66,67]; (3) postmortem brain studies have indicated reduced expression of both dysbindin-1 mRNA and protein in the brains of schizophrenic patients [22,75]; (4) dysbindin-1-deficient mice displayed schizophrenia-like behaviors, especially the negative symptoms of schizophrenia [86,87]; (five) dysbindin-1 affects the release of dopamine and glutamate and the trafficking of neurotransmitter receptors [41,46]; and (vi) dysbindin-ane is involved in neurite outgrowth [24,59,93]. Cumulatively, these findings suggest that multiple brain neuronal processes in different combinations may be affected by dysbindin-one protein expression or activity, in order to trigger schizophrenia. The evidences gathered so far indicate multiple cellular activities of dysbindin in neurons. It is quite articulate that dysbindin enters the nucleus to regulate transcription and shuttle from the nucleus to the cytoplasm to assemble into various multi-subunit protein complexes (e.k., BLOC-1) regulating the cytoskeleton, trafficking and signaling pathways. Moreover, multiple biologically active forms of dysbindin-one would control neurite outgrowth and dendritic spine maturation during neuronal differentiation and, in mature neurons, the biogenesis and release of synaptic vesicles at pre-synaptic terminals as well as the downwards-regulation of neurotransmitter receptors at postsynaptic terminals. Thus, it is imperative to further expand our cognition of multiple dysbindin-1 interacting poly peptide candidates to schizophrenia disease susceptibility. A focus but on dysbindin-1 protein is unlikely to unravel this circuitous disease. Dysbindin-one may confer its susceptibility to schizophrenia through its impact on dopaminergic and glutamatergic neurotransmission, which link to other neurochemical brain pathways. We also propose that dysbindin-1 might be a potential therapeutic target for the treatment of this schizophrenia. The mechanism(southward) by which dysbindin-1 affects dopaminergic and glutamatergic signaling, and its furnishings on the different receptor subtypes and downstream molecules need further investigations. It is besides of import to examine the impact of environmental stress on the expression of dysbindin-1 levels in both beast models and patients. Finally, the effects of antipsychotics on the expression of dysbindin-1 are not fully understood and need to exist further explored.
In conclusion, dysbindin-1 plays a broad role in the etiology of schizophrenia. Enhancing the level or action of dysbindin-1 in the brain might be beneficial for the treatment of schizophrenia. However, this disorder occurs through a complicated interaction of multiple biochemical, neurochemical, genetic and environmental risk factors of a poorly understood pathology. Under these circumstances, it is therefore important to clarify dysbindin-one-mediated neuronal functions and to develop therapeutic molecules and modalities based on this target.
Acknowledgments
This enquiry was supported past National Natural Science Foundation of China (No. 81301099, No. 31771128, and No. 31371088); Science and Engineering Planning Project of Guangdong Province (No. 2011B050200005); MYRG2016-00052-FHS from Academy of Macau; and the Science and Technology Development Fund (FDCT) of Macau (FDCT 021/2015/A1 and 016/2016/A1).
Conflicts of Involvement
The authors declare no disharmonize of interest.
References
- Wang, H.; Farhan, M.; Xu, J.; Lazarovici, P.; Zheng, W. The interest of darpp-32 in the pathophysiology of schizophrenia. Oncotarget 2017, 8, 53791–53803. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Wang, H.; Zeng, Z.; Lin, J.; Little, P.J.; Srivastava, Fifty.K.; Quirion, R. The possible role of the akt signaling pathway in schizophrenia. Brain Res. 2012, 1470, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T. Managing negative symptoms of schizophrenia: How far accept we come? CNS Drugs 2017, 31, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Schooler, N.R.; Buchanan, R.W.; Laughren, T.; Leucht, South.; Nasrallah, H.A.; Potkin, S.G.; Abi-Saab, D.; Berardo, C.G.; Bugarski-Kirola, D.; Blaettler, T.; et al. Defining therapeutic benefit for people with schizophrenia: Focus on negative symptoms. Schizophr. Res. 2015, 162, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Snyder, G.L.; Vanover, K.Due east. Dopamine targeting drugs for the treatment of schizophrenia: Past, present and future. Curr. Top. Med. Chem. 2016, 16, 3385–3403. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Grand.; Walton, Due north.Grand.; Yamada, H.; Kondo, Y.; Marek, M.J.; Tajinda, K. The bear on of genetics on future drug discovery in schizophrenia. Practiced. Opin. Drug Discov. 2017, 12, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Eyre, H.; Jacka, F.Northward.; Dodd, S.; Dean, O.; McEwen, Southward.; Debnath, M.; McGrath, J.; Maes, M.; Amminger, P.; et al. A review of vulnerability and risks for schizophrenia: Beyond the two hitting hypothesis. Neurosci. Biobehav. Rev. 2016, 65, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Attademo, L.; Bernardini, F.; Garinella, R.; Compton, M.T. Environmental pollution and take a chance of psychotic disorders: A review of the science to engagement. Schizophr. Res. 2017, 181, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wang, Y.; Li, Z.; Huang, J.; Lui, S.S.; Tan, S.P.; Yu, Ten.; Cheung, E.F.; He, K.G.; Ott, J.; et al. Heritability and familiality of neurological soft signs: Testify from healthy twins, patients with schizophrenia and not-psychotic first-degree relatives. Psychol. Med. 2016, 46, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Owens, S.F.; Picchioni, M.M.; Rijsdijk, F.V.; Stahl, D.; Vassos, E.; Rodger, A.K.; Collier, D.A.; Murray, R.M.; Toulopoulou, T. Genetic overlap between episodic retentivity deficits and schizophrenia: Results from the maudsley twin report. Psychol. Med. 2011, 41, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Kendler, Yard.S.; Diehl, S.R. The genetics of schizophrenia: A electric current, genetic-epidemiologic perspective. Schizophr. Bull. 1993, xix, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Riley, B.; Kendler, K.S. Molecular genetic studies of schizophrenia. Eur. J. Hum. Genet. 2006, fourteen, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Cardno, A.Yard.; Gottesman, I.I. Twin studies of schizophrenia: From bow-and-arrow concordances to star wars mx and functional genomics. Am. J. Med. Genet. 2000, 97, 12–17. [Google Scholar] [CrossRef]
- Tang, J.; Fan, Y.; Li, H.; Xiang, Q.; Zhang, D.F.; Li, Z.; He, Y.; Liao, Y.; Wang, Y.; He, F.; et al. Whole-genome sequencing of monozygotic twins discordant for schizophrenia indicates multiple genetic hazard factors for schizophrenia. J. Genet. Genom. 2017, 44, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; Williams, N.M.; O'Donovan, M.C. Dysbindin-1 and schizophrenia: From genetics to neuropathology. J. Clin. Investig. 2004, 113, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Prats, C.; Arias, B.; Moya-Higueras, J.; Pomarol-Clotet, Eastward.; Parellada, K.; Gonzalez-Pinto, A.; Peralta, V.; Ibanez, M.I.; Martin, M.; Fananas, L.; et al. Evidence of an epistatic effect betwixt dysbindin-1 and neuritin-ane genes on the risk for schizophrenia spectrum disorders. Eur. Psychiatry 2017, 40, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, One thousand.; Morris, D.W.; Clarke, South.; McGhee, K.A.; Schwaiger, S.; Nangle, J.M.; Garavan, H.; Robertson, I.H.; Gill, M.; Corvin, A. Variance in neurocognitive performance is associated with dysbindin-1 in schizophrenia: A preliminary study. Neuropsychologia 2007, 45, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Markov, V.; Krug, A.; Krach, South.; Jansen, A.; Eggermann, T.; Zerres, K.; Stocker, T.; Shah, N.J.; Nothen, Yard.M.; Treutlein, J.; et al. Impact of schizophrenia-adventure factor dysbindin 1 on brain activation in bilateral middle frontal gyrus during a working memory task in healthy individuals. Hum. Brain Mapp. 2010, 31, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Yang, F.; Xiao, Y.; Tan, S.; Husain, North.; Ren, G.; Hu, Z.; Martinowich, K.; Ng, J.S.; Kim, P.J.; et al. Regulation of brain-derived neurotrophic factor exocytosis and gamma-aminobutyric acidergic interneuron synapse past the schizophrenia susceptibility gene dysbindin-1. Biol. Psychiatry 2016, 80, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Northward.; Onaka, Y.; Hashimoto, R.; Takamura, H.; Nagata, T.; Umeda-Yano, S.; Mouri, A.; Mamiya, T.; Haba, R.; Matsuzaki, S.; et al. Behavioral characterization of mice overexpressing human dysbindin-i. Mol. Brain 2014, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Petit, E.I.; Michalak, Z.; Cox, R.; O'Tuathaigh, C.Yard.; Clarke, N.; Tighe, O.; Talbot, M.; Blake, D.; Joel, J.; Shaw, A.; et al. Dysregulation of specialized delay/interference-dependent working memory following loss of dysbindin-1a in schizophrenia-related phenotypes. Neuropsychopharmacoloy 2017, 42, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Talbot, Yard.; Eidem, W.Fifty.; Tinsley, C.50.; Benson, Chiliad.A.; Thompson, E.W.; Smith, R.J.; Hahn, C.G.; Siegel, Southward.J.; Trojanowski, J.Q.; Gur, R.E.; et al. Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J. Clin. Investig. 2004, 113, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Weickert, C.S.; Straub, R.E.; McClintock, B.West.; Matsumoto, One thousand.; Hashimoto, R.; Hyde, T.M.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.Due east. Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch. Gen. Psychiatry 2004, 61, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Ghiani, C.A.; Starcevic, M.; Rodriguez-Fernandez, I.A.; Nazarian, R.; Cheli, V.T.; Chan, L.N.; Malvar, J.South.; de Vellis, J.; Sabatti, C.; Dell'Angelica, E.C. The dysbindin-containing circuitous (BLOC-i) in encephalon: Developmental regulation, interaction with SNARE proteins and role in neurite outgrowth. Mol. Psychiatry 2010, xv, 115. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Fei, East.; Fu, C.; Ren, H.; Wang, Thousand. Dysbindin-1, a schizophrenia-related protein, facilitates neurite outgrowth by promoting the transcriptional activeness of p53. Mol. Psychiatry 2011, 16, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, F.; Burdick, M.C.; Callicott, J.H.; Weinberger, D.R. Epistatic interaction between comt and DTNBP1 modulates prefrontal role in mice and in humans. Mol. Psychiatry 2014, xix, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yang, F.; Papaleo, F.; Wang, H.X.; Gao, W.J.; Weinberger, D.R.; Lu, B. Office of dysbindin in dopamine receptor trafficking and cortical gaba function. Proc. Natl. Acad. Sci. United states of america 2009, 106, 19593–19598. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; LeGros, R.P.; Louneva, N.; Yeh, L.; Cohen, J.W.; Hahn, C.G.; Blake, D.J.; Arnold, S.E.; Talbot, 1000. Dysbindin-i in dorsolateral prefrontal cortex of schizophrenia cases is reduced in an isoform-specific manner unrelated to dysbindin-i mrna expression. Hum. Mol. Genet. 2009, 18, 3851–3863. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yuan, Y.; Zhang, Z.; Yan, H.; Feng, Y.; Li, Due west. Dysbindin-1c is required for the survival of hilar mossy cells and the maturation of adult newborn neurons in dentate gyrus. J. Biol. Chem. 2014, 289, 29060–29072. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.Due east.; Jiang, Y.; MacLean, C.J.; Ma, Y.; Webb, B.T.; Myakishev, M.5.; Harris-Kerr, C.; Wormley, B.; Sadek, H.; Kadambi, B.; et al. Genetic variation in the 6p22.iii cistron DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am. J. Hum. Genet. 2002, 71, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Jockusch, H. Decreased osmotic stability of dystrophin-less musculus cells from the mdx mouse. Nature 1991, 349, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Benson, 1000.A.; Newey, S.E.; Martin-Rendon, East.; Hawkes, R.; Blake, D.J. Dysbindin, a novel coiled-coil-containing protein that interacts with the dystrobrevins in muscle and brain. J. Biol. Chem. 2001, 276, 24232–24241. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Morishita, R.; Shinoda, T.; Iwamoto, I.; Sudo, K.; Okamoto, K.; Nagata, One thousand. Dysbindin-1, wave2 and abi-i class a circuitous that regulates dendritic spine germination. Mol. Psychiatry 2010, 15, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Starcevic, Yard.; Spencer, M.J.; Dell'Angelica, East.C. Reinvestigation of the dysbindin subunit of bloc-ane (biogenesis of lysosome-related organelles complex-ane) equally a dystrobrevin-bounden protein. Biochem. J. 2006, 395, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Falcon-Perez, J.M.; Starcevic, M.; Gautam, R.; Dell'Angelica, E.C. Bloc-1, a novel complex containing the pallidin and muted proteins involved in the biogenesis of melanosomes and platelet-dense granules. J. Biol. Chem. 2002, 277, 28191–28199. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Louneva, North.; Cohen, J.W.; Kazi, H.; Blake, D.J.; Arnold, S.E. Synaptic dysbindin-1 reductions in schizophrenia occur in an isoform-specific way indicating their subsynaptic location. PLoS ONE 2011, 6, e16886. [Google Scholar] [CrossRef] [PubMed]
- Larimore, J.; Ryder, P.V.; Kim, K.Y.; Ambrose, Fifty.A.; Chapleau, C.; Calfa, G.; Gross, C.; Bassell, One thousand.J.; Pozzo-Miller, L.; Smith, Y.; et al. Mecp2 regulates the synaptic expression of a dysbindin-bloc-1 network component in mouse encephalon and human induced pluripotent stalk jail cell-derived neurons. PLoS ONE 2013, 8, e65069. [Google Scholar] [CrossRef] [PubMed]
- Starcevic, M.; Dell'Angelica, E.C. Identification of snapin and three novel proteins (blos1, blos2, and blos3/reduced pigmentation) every bit subunits of biogenesis of lysosome-related organelles circuitous-1 (bloc-1). J. Biol Chem. 2004, 279, 28393–28401. [Google Scholar] [CrossRef] [PubMed]
- Dickman, D.Chiliad.; Davis, G.W. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science 2009, 326, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Newell-Litwa, K.; Salazar, G.; Smith, Y.; Faundez, V. Roles of bloc-one and adaptor protein-3 complexes in cargo sorting to synaptic vesicles. Mol. Biol. Cell 2009, 20, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, Y.; Sei, Y.; Weinberger, D.R.; Straub, R.E. Evidence that the bloc-1 poly peptide dysbindin modulates dopamine d2 receptor internalization and signaling just not d1 internalization. J. Neurosci. 2007, 27, 12390–12395. [Google Scholar] [CrossRef] [PubMed]
- Oyama, S.; Yamakawa, H.; Sasagawa, Due north.; Hosoi, Y.; Futai, Eastward.; Ishiura, S. Dysbindin-1, a schizophrenia-related protein, functionally interacts with the DNA-dependent protein kinase complex in an isoform-dependent manner. PLoS ONE 2009, 4, e4199. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, Y.; Ye, H.; Zhu, 50.; Liu, J.; Wu, 10.; Wang, L.; He, T.; Shen, Y.; Wu, J.Y.; et al. Increased dysbindin-1b isoform expression in schizophrenia and its propensity in aggresome germination. Prison cell Discov. 2015, 1, 15032. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S.; Murotani, T.; Matsuzaki, S.; Ishizuka, T.; Kumamoto, N.; Takeda, M.; Tohyama, M.; Yamatodani, A.; Kunugi, H.; Hashimoto, R. Behavioral abnormalities and dopamine reductions in sdy mutant mice with a deletion in DTNBP1, a susceptibility gene for schizophrenia. Biochem. Biophys. Res. Commun. 2008, 373, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Due west.; Feng, Y.Q.; Hao, C.J.; Guo, X.L.; He, 10.; Zhou, Z.Y.; Guo, North.; Huang, H.P.; Xiong, W.; Zheng, H.; et al. DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. J. Cell Biol. 2008, 181, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, F.; Yang, F.; Garcia, Due south.; Chen, J.; Lu, B.; Crawley, J.North.; Weinberger, D.R. Dysbindin-1 modulates prefrontal cortical activity and schizophrenia-like behaviors via dopamine/d2 pathways. Mol. Psychiatry 2012, 17, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Metzinger, L.; Blake, D.J.; Squier, M.V.; Anderson, 50.Five.; Deconinck, A.Eastward.; Nawrotzki, R.; Hilton-Jones, D.; Davies, K.E. Dystrobrevin deficiency at the sarcolemma of patients with muscular dystrophy. Hum. Mol. Genet. 1997, 6, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Selemon, L.D.; Zecevic, N. Schizophrenia: A tale of 2 critical periods for prefrontal cortical development. Transl. Psychiatry 2015, v, e623. [Google Scholar] [CrossRef] [PubMed]
- Soma, M.; Wang, M.; Suo, S.; Ishiura, S. Dysbindin-1, a schizophrenia-related protein, interacts with hdac3. Neurosci. Lett. 2014, 582, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Chen, D.; Chen, R.; Hu, Q.; Wang, Grand. The schizophrenia-related protein dysbindin-1a is degraded and facilitates nf-kappa b action in the nucleus. PLoS ONE 2015, ten, e0132639. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.O.M. Dual constraints on synapse formation and regression in schizophrenia: Neuregulin, neuroligin, dysbindin, disc1, musk and agrin. Aust. Due north. Z. J. Psychiatry 2008, 42, 662–677. [Google Scholar] [CrossRef] [PubMed]
- Fei, E.; Ma, X.; Zhu, C.; Xue, T.; Yan, J.; Xu, Y.; Zhou, J.; Wang, 1000. Nucleocytoplasmic shuttling of dysbindin-i, a schizophrenia-related protein, regulates synapsin i expression. J. Biol. Chem. 2010, 285, 38630–38640. [Google Scholar] [CrossRef] [PubMed]
- Cha, D.S.; Kudlow, P.A.; Baskaran, A.; Mansur, R.B.; McIntyre, R.Due south. Implications of epigenetic modulation for novel treatment approaches in patients with schizophrenia. Neuropharmacology 2014, 77, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Boersma, Chiliad.C.; Dresselhaus, E.C.; De Biase, L.M.; Mihalas, A.B.; Bergles, D.E.; Meffert, Thousand.G. A requirement for nuclear factor-kappab in developmental and plasticity-associated synaptogenesis. J. Neurosci. 2011, 31, 5414–5425. [Google Scholar] [CrossRef] [PubMed]
- Roussos, P.; Katsel, P.; Davis, 1000.L.; Giakoumaki, South.G.; Lencz, T.; Malhotra, A.K.; Siever, 50.J.; Bitsios, P.; Haroutunian, V. Convergent findings for abnormalities of the nf-kappab signaling pathway in schizophrenia. Neuropsychopharmacology 2013, 38, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, Grand.; Diniz, B.S.; Leszek, J. Inflammatory response in the CNS: Friend or foe? Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Culmsee, C.; Yu, Z.; Camandola, Due south. Roles of nuclear factor kappab in neuronal survival and plasticity. J. Neurochem. 2000, 74, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, S.D.; Juczewski, Yard.; de Haan, A.M.; Seaton, G.; Play a joke on, Chiliad.; Hardingham, N.R. Adult cortical plasticity depends on an early postnatal critical catamenia. Science 2015, 349, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Kim, S.Chiliad.; Suh, B.1000.; Dominicus, H.Y.; Park, Y.U.; Hong, J.H.; Park, C.; Nguyen, One thousand.D.; Nagata, K.; Yoo, J.Y.; et al. Disrupted-in-schizophrenia 1 (disc1) regulates dysbindin function past enhancing its stability. J. Biol. Chem. 2015, 290, 7087–7096. [Google Scholar] [CrossRef] [PubMed]
- Talbot, G.; Cho, D.S.; Ong, West.Y.; Benson, 1000.A.; Han, L.Y.; Kazi, H.A.; Kamins, J.; Hahn, C.Grand.; Blake, D.J.; Arnold, Due south.E. Dysbindin-1 is a synaptic and microtubular protein that binds brain snapin. Hum. Mol. Genet. 2006, fifteen, 3041–3054. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, J.1000.; Mochida, S.; Sheng, Z.H. Snapin: A snare-associated protein implicated in synaptic manual. Nat. Neurosci. 1999, 2, 119–124. [Google Scholar] [PubMed]
- Feng, Y.Q.; Zhou, Z.Y.; He, X.; Wang, H.; Guo, X.L.; Hao, C.J.; Guo, Y.; Zhen, 10.C.; Li, W. Dysbindin deficiency in sandy mice causes reduction of snapin and displays behaviors related to schizophrenia. Schizophr. Res. 2008, 106, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Farrell, Thou.S.; Werge, T.; Sklar, P.; Owen, Chiliad.J.; Ophoff, R.A.; O'Donovan, G.C.; Corvin, A.; Cichon, S.; Sullivan, P.F. Evaluating historical candidate genes for schizophrenia. Mol. Psychiatry 2015, 20, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Schwab, S.Thou.; Knapp, M.; Mondabon, Due south.; Hallmayer, J.; Borrmann-Hassenbach, M.; Albus, M.; Lerer, B.; Rietschel, M.; Trixler, G.; Maier, W.; et al. Support for association of schizophrenia with genetic variation in the 6p22.3 gene, dysbindin, in sib-pair families with linkage and in an boosted sample of triad families. Am. J. Hum. Genet. 2003, 72, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Cheah, S.Y.; Lawford, B.R.; Young, R.M.; Morris, C.P.; Voisey, J. Dysbindin (DTNBP1) variants are associated with hallucinations in schizophrenia. Eur. Psychiatry J. Assoc. Eur. Psychiatr. 2015, 30, 486–491. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bakanidze, M.; Brandl, East.J.; Hutzler, C.; Aurass, F.; Onken, S.; Rapp, Chiliad.A.; Puls, I. Clan of dystrobrevin-binding poly peptide one polymorphisms with sustained attention and set-shifting in schizophrenia patients. Neuropsychobiology 2016, 74, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Kim, J.S.; Ryu, S.; Oh, Due south.; Noh, J.; Lee, Due west.K.; Park, T.; Lee, Y.Southward.; Lee, D.; Kwon, J.S.; et al. Association of genetic variations in DTNBP1 with cognitive function in schizophrenia patients and healthy subjects. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Trost, Due south.; Platz, B.; Conductor, J.; Scherk, H.; Wobrock, T.; Ekawardhani, S.; Meyer, J.; Reith, W.; Falkai, P.; Gruber, O. The DTNBP1 (dysbindin-1) gene variant rs2619522 is associated with variation of hippocampal and prefrontal gray matter volumes in humans. Eur. Curvation. Psychiatry Clin. Neurosci. 2013, 263, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Voisey, J.; Swagell, C.D.; Hughes, I.P.; Connor, J.P.; Lawford, B.R.; Young, R.One thousand.; Morris, C.P. A polymorphism in the dysbindin gene (DTNBP1) associated with multiple psychiatric disorders including schizophrenia. Behav. Brain Funct. 2010, 6, 41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar][Light-green Version]
- Cox, M.Thou.; Tucker, A.M.; Tang, J.; Talbot, K.; Richer, D.C.; Yeh, L.; Arnold, S.East. Neurobehavioral abnormalities in the dysbindin-1 mutant, sandy, on a c57bl/6j genetic background. Genes Brain Behav. 2009, 8, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Saggu, Southward.; Cannon, T.D.; Jentsch, J.D.; Lavin, A. Potential molecular mechanisms for decreased synaptic glutamate release in dysbindin-1 mutant mice. Schizophr. Res. 2013, 146, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, South.Yard.; Ryan, R.T.; Wong, T.P.; Srivastava, L.M. Loss of dysbindin-one, a take a chance gene for schizophrenia, leads to dumb group i metabotropic glutamate receptor function in mice. Front. Behav. Neurosci. 2015, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, F.; Weinberger, D.R. Dysbindin and schizophrenia: It'south dopamine and glutamate all over once more. Biol. Psychiatry 2011, 69, two–4. [Google Scholar] [CrossRef] [PubMed]
- Kendler, K.Due south. Schizophrenia genetics and dysbindin: A corner turned? Am. J. Psychiatry 2004, 161, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K. The sandy (sdy) mouse: A dysbindin-1 mutant relevant to schizophrenia research. Prog. Brain Res. 2009, 179, 87–94. [Google Scholar] [PubMed]
- Takao, Thou.; Toyama, K.; Nakanishi, K.; Hattori, S.; Takamura, H.; Takeda, Grand.; Miyakawa, T.; Hashimoto, R. Impaired long-term retention retention and working retention in sdy mutant mice with a deletion in DTNBP1, a susceptibility gene for schizophrenia. Mol. Brain 2008, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T. Schizophrenia: Basic and clinical. Adv. Neurobiol. 2017, 15, 255–280. [Google Scholar] [PubMed]
- Carr, G.5.; Jenkins, K.A.; Weinberger, D.R.; Papaleo, F. Loss of dysbindin-1 in mice impairs advantage-based operant learning by increasing impulsive and compulsive behavior. Behav. Encephalon Res. 2013, 241, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Braff, D.L.; Geyer, Grand.A.; Swerdlow, N.R. Human studies of prepulse inhibition of startle: Normal subjects, patient groups, and pharmacological studies. Psychopharmacology 2001, 156, 234–258. [Google Scholar] [CrossRef] [PubMed]
- Karlsgodt, K.H.; Robleto, K.; Trantham-Davidson, H.; Jairl, C.; Cannon, T.D.; Lavin, A.; Jentsch, J.D. Reduced dysbindin expression mediates n-methyl-d-aspartate receptor hypofunction and impaired working memory performance. Biol. Psychiatry 2011, 69, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A. A dual hitting model for dopamine in schizophrenia. Biol. Psychiatry 2017, 81, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.J.; Chohan, Chiliad.O.; Slifstein, 1000.; Kegeles, L.S.; Moore, H.; Abi-Dargham, A. Pathway-specific dopamine abnormalities in schizophrenia. Biol. Psychiatry 2017, 81, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Horga, 1000.; Cassidy, C.M.; Xu, X.; Moore, H.; Slifstein, M.; Van Snellenberg, J.X.; Abi-Dargham, A. Dopamine-related disruption of functional topography of striatal connections in unmedicated patients with schizophrenia. JAMA Psychiatry 2016, 73, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Schmieg, Due north.; Rocchi, C.; Romeo, S.; Maggio, R.; Millan, M.J.; Mannoury la Cour, C. Dysbindin-1 modifies signaling and cellular localization of recombinant, human d(iii) and d(2) receptors. J. Neurochem. 2016, 136, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.T.; Yang, F.; Chen, B.S.; Lu, Y.; Ji, Y.; Roche, K.W.; Lu, B. Dysbindin regulates hippocampal ltp by controlling nmda receptor surface expression. Proc. Natl. Acad. Sci. United states 2009, 106, 21395–21400. [Google Scholar] [CrossRef] [PubMed]
- Glen, Westward.B., Jr.; Horowitz, B.; Carlson, One thousand.C.; Cannon, T.D.; Talbot, One thousand.; Jentsch, J.D.; Lavin, A. Dysbindin-1 loss compromises nmdar-dependent synaptic plasticity and contextual fear conditioning. Hippocampus 2014, 24, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, J.D.; Trantham-Davidson, H.; Jairl, C.; Tinsley, M.; Cannon, T.D.; Lavin, A. Dysbindin modulates prefrontal cortical glutamatergic circuits and working memory function in mice. Neuropsychopharmacology 2009, 34, 2601–2608. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kulhara, P. What is schizophrenia: A neurodevelopmental or neurodegenerative disorder or a combination of both? A critical assay. Indian J. Psychiatry 2010, 52, 21–27. [Google Scholar] [PubMed]
- Lefebvre, J.L.; Sanes, J.R.; Kay, J.N. Development of dendritic form and office. Annu. Rev. Jail cell Dev. Biol. 2015, 31, 741–777. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Kumamoto, Northward.; Matsuzaki, S.; Hashimoto, R.; Hattori, T.; Okuda, H.; Takamura, H.; Takeda, M.; Katayama, T.; Tohyama, M. Dysbindin engages in c-jun n-terminal kinase activity and cytoskeletal organization. Biochem. Biophys. Res. Commun. 2009, 379, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Tomoda, T.; Chang, J.; Takaki, M.; Zhan, C.; Morita, M.; Cascio, Thou.B.; Elashvili, S.; Koizumi, H.; Takanezawa, Y.; et al. Disc1-ndel1/nudel poly peptide interaction, an essential component for neurite outgrowth, is modulated by genetic variations of disc1. Hum. Mol. Genet. 2006, xv, 3313–3323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yan, Westward.; Chen, X. P53 is required for nerve growth factor-mediated differentiation of pc12 cells via regulation of trka levels. Cell Death Differ. 2006, xiii, 2118–2128. [Google Scholar] [CrossRef] [PubMed]
- Ryder, P.Five.; Faundez, V. Schizophrenia: The "bloc" may be in the endosomes. Sci. Signal 2009, two, pe66. [Google Scholar] [CrossRef] [PubMed]
- Mead, C.L.; Kuzyk, Grand.A.; Moradian, A.; Wilson, G.G.; Holt, R.A.; Morin, Thou.B. Cytosolic protein interactions of the schizophrenia susceptibility gene dysbindin. J. Neurochem. 2010, 113, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C. The synaptic vesicle bicycle. Annu. Rev. Neurosci. 2004, 27, 509–547. [Google Scholar] [CrossRef] [PubMed]
- Hikita, T.; Taya, S.; Fujino, Y.; Taneichi-Kuroda, South.; Ohta, K.; Tsuboi, D.; Shinoda, T.; Kuroda, K.; Funahashi, Y.; Uraguchi-Asaki, J.; et al. Proteomic assay reveals novel binding partners of dysbindin, a schizophrenia-related protein. J. Neurochem. 2009, 110, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, A.; Hartwig, C.; Freeman, A.H.; Das, R.; Zlatic, S.A.; Vistein, R.; Burch, A.; Carrot, G.; Lewis, A.F.; Nelms, S.; et al. The proteome of bloc-ane genetic defects identifies the arp2/3 actin polymerization complex to function downstream of the schizophrenia susceptibility factor dysbindin at the synapse. J. Neurosci. 2016, 36, 12393–12411. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.H.; Racz, B.; Wang, H.; Burianek, L.; Weinberg, R.; Yasuda, R.; Wetsel, W.C.; Soderling, S.H. Disruption of arp2/iii results in disproportionate structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J. Neurosci. 2013, 33, 6081–6092. [Google Scholar] [CrossRef] [PubMed]
- Rocca, D.Fifty.; Amici, M.; Antoniou, A.; Blanco Suarez, Due east.; Halemani, N.; Murk, Chiliad.; McGarvey, J.; Jaafari, North.; Mellor, J.R.; Collingridge, G.50.; et al. The pocket-sized gtpase arf1 modulates arp2/three-mediated actin polymerization via pick1 to regulate synaptic plasticity. Neuron 2013, 79, 293–307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jia, J.M.; Hu, Z.; Nordman, J.; Li, Z. The schizophrenia susceptibility gene dysbindin regulates dendritic spine dynamics. J. Neurosci. 2014, 34, 13725–13736. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, A.; Mullin, A.P.; Zlatic, S.A.; Easley, C.A.; Merritt, M.Due east.; Raj, N.; Larimore, J.; Gordon, D.E.; Peden, A.A.; Sanyal, Southward.; et al. The north-ethylmaleimide-sensitive cistron and dysbindin interact to modulate synaptic plasticity. J. Neurosci. 2015, 35, 7643–7653. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, A.; Larimore, J.; Werner, E.; Then, 50.; Moreno-De-Luca, A.; Lese-Martin, C.; Lupashin, V.Five.; Smith, Y.; Faundez, V. Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex i. J. Neurosci. 2012, 32, 3697–3711. [Google Scholar] [CrossRef] [PubMed]
- Han, M.H.; Hu, Z.; Chen, C.Y.; Chen, Y.; Gucek, M.; Li, Z.; Markey, S.P. Dysbindin-associated proteome in the p2 synaptosome fraction of mouse brain. J. Proteome Res. 2014, 13, 4567–4580. [Google Scholar] [CrossRef] [PubMed]
- Guillozet-Bongaarts, A.L.; Hyde, T.M.; Dalley, R.A.; Hawrylycz, K.J.; Henry, A.; Hof, P.R.; Hohmann, J.; Jones, A.R.; Kuan, C.50.; Royall, J.; et al. Altered gene expression in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 2014, 19, 478–485. [Google Scholar] [CrossRef] [PubMed]
Figure i. Interaction of dysbindin-1 with cellular proteins. Dysbindin-i has been shown to associate into complexes with multiple binding partners, in both the cytoplasm and nucleus. Dysbindin-ane binds to α- and β-dystrobrevins and regulates muscular and neuronal morphologies. Dysbindin-1 may likewise collaborate with snapin and disrupted in schizophrenia 1 (DISC1), and thereby modulating neurotransmitter release and neurodevelopment, respectively. In the nucleus, dysbindin-1 can class a circuitous with Dna-dependent poly peptide kinase (Dna-PK) and promote the phosphorylation of histone deacetylase 3 (HDAC3). In improver, interaction of dysbindin-1 with DISC1 may dysregulate neuronal development.
Figure ane. Interaction of dysbindin-i with cellular proteins. Dysbindin-i has been shown to acquaintance into complexes with multiple binding partners, in both the cytoplasm and nucleus. Dysbindin-1 binds to α- and β-dystrobrevins and regulates muscular and neuronal morphologies. Dysbindin-1 may also interact with snapin and disrupted in schizophrenia 1 (DISC1), and thereby modulating neurotransmitter release and neurodevelopment, respectively. In the nucleus, dysbindin-ane can form a circuitous with Dna-dependent poly peptide kinase (Deoxyribonucleic acid-PK) and promote the phosphorylation of histone deacetylase 3 (HDAC3). In addition, interaction of dysbindin-1 with DISC1 may dysregulate neuronal development.
Effigy 2. Regulation of dysbindin-ane on pre-synaptic vesicles and mail service-synaptic receptors. In the pre-synaptic terminals, dysbindin-ane regulates synaptic vesicle transport and release. In the post synaptic terminals, dysbindin-i regulates the trafficking of neurotransmitter receptors, such as dopaminergic receptors, and affects the expression of these receptors on neuronal cell surface. Dysbindin-1 also modifies protein kinase B (Akt) and extracellular signal-regulated kinase1/2 (ERK1/ii) signaling through acting on M poly peptide receptor-induced adenylate cyclase recruitment and army camp production.
Figure 2. Regulation of dysbindin-1 on pre-synaptic vesicles and mail service-synaptic receptors. In the pre-synaptic terminals, dysbindin-1 regulates synaptic vesicle transport and release. In the post synaptic terminals, dysbindin-one regulates the trafficking of neurotransmitter receptors, such as dopaminergic receptors, and affects the expression of these receptors on neuronal jail cell surface. Dysbindin-1 besides modifies poly peptide kinase B (Akt) and extracellular point-regulated kinase1/two (ERK1/2) signaling through interim on Chiliad protein receptor-induced adenylate cyclase recruitment and camp production.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access commodity distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Source: https://www.mdpi.com/1422-0067/18/10/2044/htm
{kind=link}
Post a Comment for "Dysbindin and Schizophrenia: Itã¢â‚¬â„¢s Dopamine and Glutamate All Over Again."